Wave Function Ansatz in QMCTorch
QMCTorch allows to epxress the wave function ususally used by QMC practitioner as neural network. The most generic architecture of the neural network used by the code is:
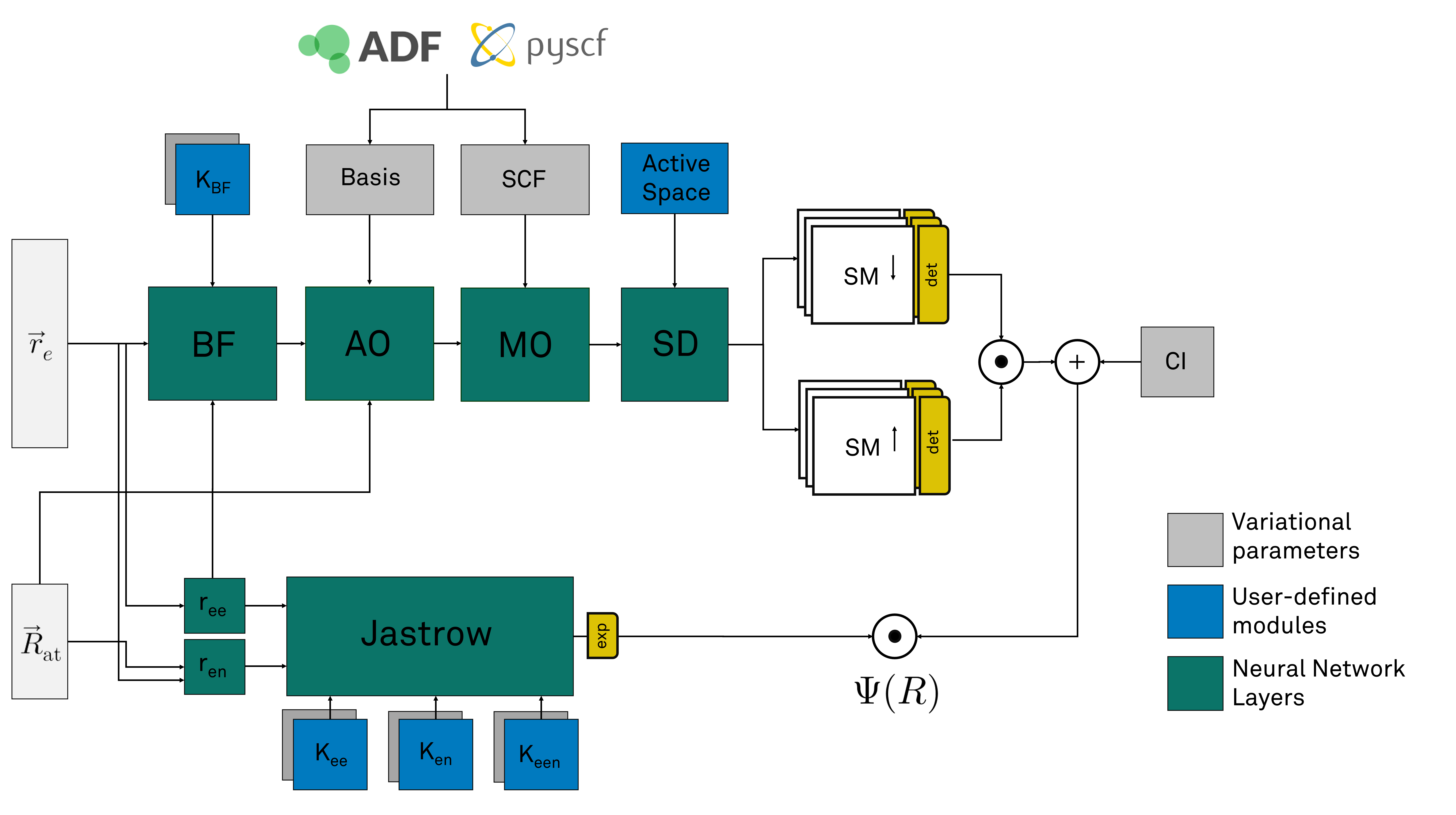
Starting from the electronic and atomic coordinates, the first layer on the bottom computes the electron-electron and electron-atoms distances. These distances are used in a Jastrow layer that computes the Jastrow facrtor. Users can freely define Jastrow kernels to define the exact form the Jastrow factor.
In parallel the electronic coordinates are first transformed through a backflow transformation. Users can here as well specify the kernel of the backflow transformation.
The resulting new coordinates are used to evaluate the atomic orbitals of the systems. The basis set information of these orbitals are extracted from the SCF calculation performed with pyscf or ADF.
These atomic orbital values are then transformed to molecular orbital values through the next layer. The coefficients of the molecular orbitals are also extracted fron the SCF calculations.
Then a Slater determinant layer extract the different determinants contained in the wave function. Users can there as well specify wich determinants they require. The weighted sum of the determinants
is then computed and finally muliplied with the value of the Jastrow factor.
The main wave function in QMCTorch is implemented in the SlaterJastrow class. The definition of the class is as follows :
class SlaterJastrow(WaveFunction):
def __init__(
self,
mol,
jastrow='default',
backflow=None,
configs="ground_state",
kinetic="jacobi",
cuda=False,
include_all_mo=True,
):
Different functional form can be created from this class depending on the need of the user. We review here a few of these forms.
Simple Slater Jastrow Wave Function
The simplest wave function implemented in QMCTorch is a Slater Jastrow form. Through a series of transformations the Slater Jastrow function computes:
The term J(R) is the so called Jastrow factor that captures the electronic correlation. By default, the Jastrow factor is given by :
where the sum runs over all the electron pairs and where the kernel function defines the action of the Jastrow factor. A common expression for the kernel (and the default option for QMCTorch) is the Pade-Jastrow form given by:
where \(\omega_0\) is a fixed coefficient equals to 0.25(0.5) for antiparallel(parallel) electron spins and \(\omega\) a variational parameter.
The determinantal parts in the expression of \(\Psi\) are given by the spin-up and spin-down slater determinants e.g. :
A SlaterJastrow wave function can instantiated following :
from qmctorch.scf import Molecule
from qmctorch.wavefunction.slater_jastrow import SlaterJastrow
from qmctorch.wavefunction.jastrows.elec_elec.jastrow_factor_electron_electron import JastrowFactorElectronElectron
from qmctorch.wavefunction.jastrows.elec_elec.kernels import PadeJastrowKernel
mol = Molecule('H 0 0 0; H 0 0 1')
jastrow = JastrowFactorElectronElectron(mol, PadeJastrowKernel)
wf = SlaterJastrow(mol, configs='single_double(2,2)', jastrow=jastrow)
The SlaterJastrow takes as first mandiatory argument a Molecule instance. The Slater determinants required in the calculation
are specified with the configs arguments which can take the following values :
configs='ground_state': only the ground state SD
configs='cas(n,m)': complete active space using n electron and m orbitals
configs='single(n,m)': only single excitation using n electron and m orbitals
configs='single_double(n,m)': only single/double excitation using n electron and m orbitals
Finally the Jastrow factor can be specifed using the jastrow. We used here a Pade-Jastrow kernel that is already implemented in QMCTorch
Custom Jastrow factor
It is possible to define custom Jastrow factor and use these forms in the definition of the wave function.
from torch import nn
from qmctorch.wavefunction import SlaterJastrow
from qmctorch.wavefunction.jastrows.elec_elec.jastrow_factor_electron_electron import JastrowFactorElectronElectron
from qmctorch.wavefunction.jastrows.elec_elec.kernels import JastrowKernelElectronElectronBase
class MyJastrowKernel(JastrowKernelElectronElectronBase):
def __init__(self, nup, ndown, cuda, size=16):
super().__init__(nup, ndown, cuda)
self.fc1 = nn.Linear(1, size, bias=False)
self.fc2 = nn.Linear(size, 1, bias=False)
def forward(self, x):
nbatch, npair = x.shape
x = x.reshape(-1,1)
x = self.fc2(self.fc1(x))
return x.reshape(nbatch, npair)
mol = Molecule(atom='H 0. 0. 0; H 0. 0. 1.', calculator='pyscf', unit='bohr', redo_scf=True)
jastrow = JastrowFactorElectronElectron(mol, MyJastrowKernel, kernel_kwargs={'size': 64})
wf = SlaterJastrow(mol, jastrow=jastrow)
Combining Several Jastrow Factors
As shown on the figure above it is possible to combine several Jastrow factors to account for not only the electron-electron correlations but also electron-nuclei and three body terms. This can easily be done by passing a list of Jastrow factors to the SlaterJastrow wave function. For example if we want to combine a fully connected electron-electron neural Jastrow factor with a fully connected electron-nuclei neural Jastrow, we can simply use:
import torch
from qmctorch.scf import Molecule
from qmctorch.wavefunction import SlaterJastrow
from qmctorch.wavefunction.jastrows.elec_elec import (
JastrowFactor as JastrowFactorElecElec,
FullyConnectedJastrowKernel as FCEE,
)
from qmctorch.wavefunction.jastrows.elec_nuclei import (
JastrowFactor as JastrowFactorElecNuclei,
FullyConnectedJastrowKernel as FCEN,
)
mol = Molecule(
atom="Li 0 0 0; H 0 0 3.14",
unit='bohr',
calculator="pyscf",
basis="sto-3g",
redo_scf=True)
jastrow_ee = JastrowFactorElecElec(mol, FCEE)
jastrow_en = JastrowFactorElecNuclei(mol, FCEN)
wf = SlaterJastrow(mol, jastrow=[jastrow_ee, jastrow_en])
Wave Functions with Backflow Transformations
As seen on the figure above, a backflow transformation of the electronic positions can be added to the definition of the wave function. Following this transformation, each electron becomes a quasi-particle whose position depends on all electronic positions. The backflow transformation is given by :
The kernel of the transformation can be any function that depends on the distance between two electrons. A popular kernel is simply the inverse function :
and is the default value in QMCTorch. However any other kernel function can be implemented and used in the code.
The wave function is then constructed as :
The Jastrow factor is still computed using the original positions of the electrons while the determinant part uses the backflow transformed positions. One can define such wave function with:
from qmctorch.scf import Molecule
from qmctorch.wavefunction.slater_jastrow import SlaterJastrow
from qmctorch.wavefunction.jastrows.elec_elec import JastrowFactor, PadeJastrowKernel
from qmctorch.wavefunction.orbitals.backflow import (
BackFlowTransformation,
BackFlowKernelInverse,
)
# molecule
mol = Molecule(
atom="Li 0 0 0; H 0 0 3.015",
unit="bohr",
calculator="pyscf",
basis="sto-3g",
redo_scf=True,
)
# define jastrow factor
jastrow = JastrowFactor(mol, PadeJastrowKernel)
# define backflow trans
backflow = BackFlowTransformation(mol, BackFlowKernelInverse)
# define the wave function
wf = SlaterJastrow(
mol,
configs="single_double(2,2)",
jastrow=jastrow,
backflow=backflow,
)
Custom Backflow Transformation
As for the Jastrow factor, it is possible to create custom backlfow transformations and use them in the definition of the wave function. For example to define a fully connected backflow kernel and use it we can use:
import torch
from torch import nn
from qmctorch.scf import Molecule
from qmctorch.wavefunction import SlaterJastrow
from qmctorch.wavefunction.orbitals.backflow.kernels import BackFlowKernelBase
from qmctorch.wavefunction.orbitals.backflow import BackFlowTransformation
class MyBackflowKernel(BackFlowKernelBase):
def __init__(self, mol, cuda, size=16):
super().__init__(mol, cuda)
self.fc1 = nn.Linear(1, size, bias=False)
self.fc2 = nn.Linear(size, 1, bias=False)
def forward(self, x):
original_shape = x.shape
x = x.reshape(-1,1)
x = self.fc2(self.fc1(x))
return x.reshape(*original_shape)
mol = Molecule(atom='H 0. 0. 0; H 0. 0. 1.', unit='bohr', redo_scf=True)
backflow = BackFlowTransformation(mol, MyBackflowKernel, backflow_kernel_kwargs={'size': 8})
wf = SlaterJastrow(mol, backflow=backflow)